DNA Sequencing Troubleshooting Guide
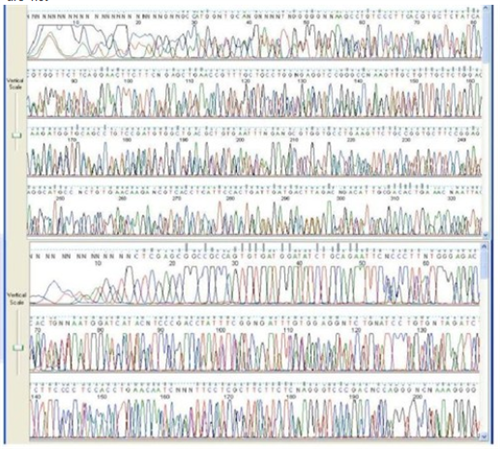
Successful DNA Sequencing Read
Peaks are well formed and separated with good quality scores. There is a small area at the beginning of the run before the chemistry stabilizes.
Examples:
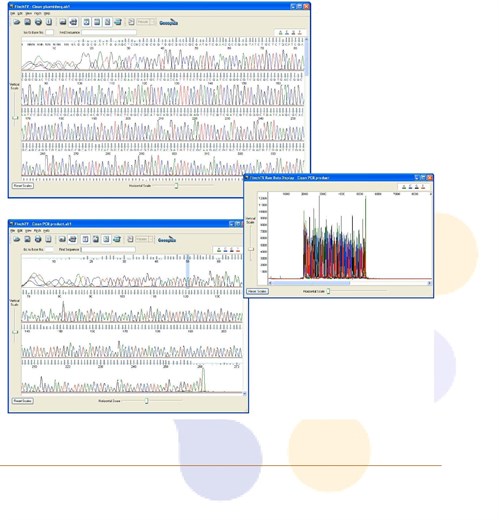
1. Failed DNA Sequencing Reaction or “Dirty” Sequence
Sequence Appearance:
- Chromatogram data looks messy or is mostly blank.
- Many "N's" in the sequence, if bases are called at all.
- Blast search from .seq or fasta file yields unexpected results.
- Raw data has signal intensity in the low hundreds.
Examples:
Possible Causes:
It is difficult to pinpoint a specific cause for the failure when there are no data for review. Common causes are:
- DNA Template concentration is too low – Note: measuring DNA concentration by UV absorption is often inaccurate and concentration is frequently overestimated. Agarose gels are a better method to estimate quality and quantity of DNA samples.
- Wrong primer or no primer was added to the reaction – binding between the DNA and the primer cannot occur in either of these instances.
- Low quality prep – yields incomplete removal of protein and/or RNA, or buffer salts or other chemical contamination may be present. Any of these conditions can inhibit sequencing reaction enzymes.
Treatment:
- Check the concentration of your template by agarose gel to ensure that it falls with the ranges listed in the Sample Submission Guidelines.
- Check the primer sequence against the template sequence to ensure that there is a proper building site.
a) Prepare fresh stock options for the template prep and dilution just prior to submitting samples.
b) Prepare plasmid DNA using a commercial mini-prep kit.
c) A final ethanol precipitation after the prep may help to ensure success.
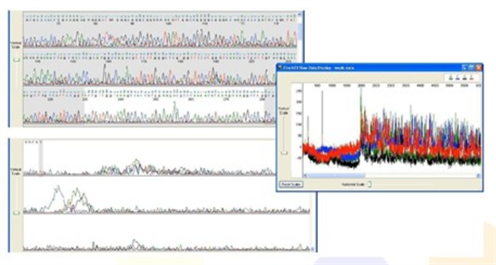
2. Double Sequence Data
Sequence Appearance:
- Sequencing peaks are not Multiple peaks with the same height or of differing heights, overlapping one another.
- Raw data has adequate signal intensity, indicating that the double peaks are not due to weak signal and/or background noise.
Possible Causes:
- Clone contamination – in this case, the beginning of the sequence is often clean and becomes "dirty‟ as the progresses past the clone insertion site.
- PCR template may be heterozygous due to indels present in a diploid (or polyploid) organism.
- Two primers may have been mistakenly added to the sequencing reaction.
- PCR products were not purified (or the purification was not performed properly). In this case, residual PCR primers may participate in the sequencing reaction.
- There may be enough homology in another area of the template that where the sequencing primer begins extension.
Treatment:
- If clone contamination is expected, please return to your clone resource and replate the bacteria onto selection media. You may want to select up to 12 clearly separated clones for sequencing to ensure you find the actual clone of interest.
- Where PCR template heterozygosity is suspected, examination of the some indication of the area of heterozygosity. Newly designed PCR primers that yield shorter products may allow you to discern where the troublesome area is. Alternatively, you may decide to redesign the sequencing primer close to the area where the problem first arises hoping to find a primer that will sequence one of the product species. If the PCR product is large, subcloning the template into shorter pieces may also provide a strategy for discerning the true sequence of the area of interest.
- When you suspect that two different primers may have been added to the reaction, you can simply repeat the reaction. If the working solution for the sequencing primer may be contaminated then going back to your original uncontaminated stock solution to prepare a new working solution or resynthesize the primer.
- Check your PCR purification protocol and the solutions used in the clean-up reaction.
- Check the primer sequence against the template sequence to ensure that there is a single binding site. Where sequence is unknown, you may need to switch to a different sequencing primer to eliminate the problem.
3. Noisy Background
Sequence Appearance
Background noise and odd peaks are present underneath the main sequence peaks.
Example:
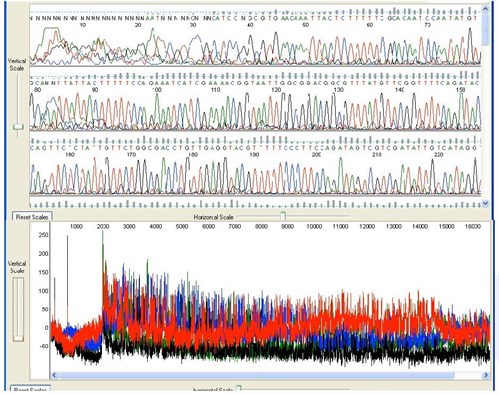
Possible Causes:
- Clone contamination – in this case, the beginning of the sequence is often clean and becomes "dirty‟ as the sequence progresses past the clone insertion site.
- PCR template may be heterozygous due to indels present in a diploid (or polyploid) organism.
- Two primers may have been mistakenly added to the sequencing reaction.
- PCR products were not purified (or the purification was not performed properly). In this case, residual PCR primers may participate in the sequencing reaction.
- There may be enough homology in another area of the template that where the sequencing primer begins extension.
- If clone contamination is expected, please return to your clone resource and replate the bacteria onto selection media. You may want to select up to 12 clearly separated clones for sequencing to ensure you find the actual clone of interest.
- Where PCR template heterozygosity is suspected, examination of the some indication of the area of heterozygosity. Newly designed PCR primers that yield shorter products may allow you to discern where the troublesome area is. Alternatively, you may decide to redesign the sequencing primer close to the area where the problem first arises hoping to find a primer that will sequence one of the product species. If the PCR product is large, subcloning the template into shorter pieces may also provide a strategy for discerning the true sequence of the area of interest.
- When you suspect that two different primers may have been added to the reaction, you can simply repeat the reaction. If the working solution for the sequencing primer may be contaminated then going back to your original uncontaminated stock solution to prepare a new working solution or resynthesize the primer.
- Check your PCR purification protocol and the solutions used in the clean-up reaction.
- Check the primer sequence against the template sequence to ensure that there is a single binding site. Where sequence is unknown, you may need to switch to a different sequencing primer to eliminate the problem.
4. Stuttering after Mononucleotide Stretches
Sequence Appearance
Sequencing data quality is poor after stretches of 7 or more nucleotides of the same base.
Example:
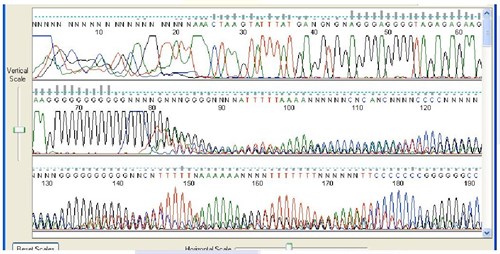
Possible Causes:
- Polymerase slippage during DNA synthesis. This is a recognized limitation of the Sanger method.
Treatment:
Options to go around the stuttering artifact are:
- Sequence from the reverse direction.
- Use a poly-mononucleotide primer with a degenerate base (wobble) at the 3′ end of the primer.
5. Mid Sequence Stop or Drop-Off
Sequence Appearance:
The DNA sequence suddenly stops or peak intensity drops off substantially. NOTE: Many popular cloning vectors have palindromes flanking their linker and may show the drop-off effect when sequenced. This is a limitation of the Sanger method, but it can many times be overcome with PowerRead technology.
Example:
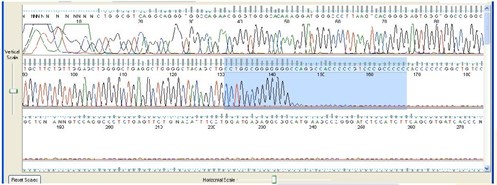
Possible Causes:
- Secondary structures in the DNA template (e.g. hairpin loops, palindromes)
- GC or GT rich regions
- Sample is an siRNA construct
Treatment:
- Power Read technology yields excellent results with these template types.
6. Top-Heavy Sequences
Sequence Appearance
Sequencing signal gradually drops off
Example:
Possible Causes:
- Excess DNA template or primer.
- GC or GT rich template (from bisulfide treated DNA, for example)
Treatment:
- Carefully quantify your DNA template and primer prior to sequencing.
- Sequence from the reverse direction.
- Use a poly-mononucleotide primer with a degenerate base (wobble) at the
7. Spikes
Sequence Appearance:
Sharp, high-intensity, multicolored peaks that randomly appear in the sequence
Example:
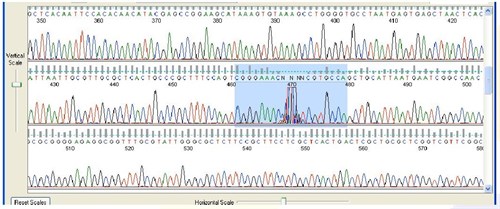
Possible Causes:
- The cause of spikes is not completely understood. One possible explanation is that an impurity or micro bubble passes through the camera view of the sequencing instrument, scattering light.
Treatment:
- The surrounding sequence should still be correct and accurate if of good quality, please request your sample to be rerun if necessary.
8. Early Peak Deterioration
Sequence Appearance
Peaks become broad or oddly shaped very early on in the sequence.
Example:
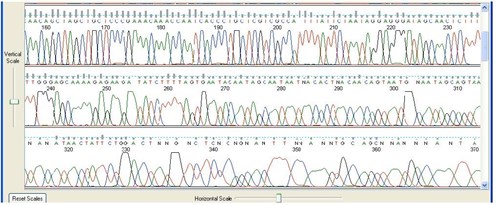
Possible Causes
- Salt or other impurities carried over from DNA isolation protocols, protocols.
Treatment
- Use a commercial desalting kit to further purify the DNA template before sequencing.
9. CRISPR and TIDE Assays
Customers that are using sequencing in an experiment related to TIDE assays should email genomicssupport@eurofins.com after submitting their order. Due to certain equipment protocols, TIDE and CRISPR related sequences can cause issues during the sequencing run. It is not an issue if we are notified early on and can adjust the protocol to match your application.